
Sequencing technologies are essential tools in biomedical research, enabling the exploration of genetic and molecular mechanisms underlying diseases. By decoding DNA and RNA sequences, researchers can identify mutations, genetic variations, and gene expression patterns, which contribute to better understanding, diagnosis, and treatment of various diseases. From whole-genome sequencing (WGS) to RNA-sequencing (RNA-seq), these technologies help reveal the genetic basis of disorders, allowing for targeted therapies and personalized medicine.
Transcriptomic Analysis
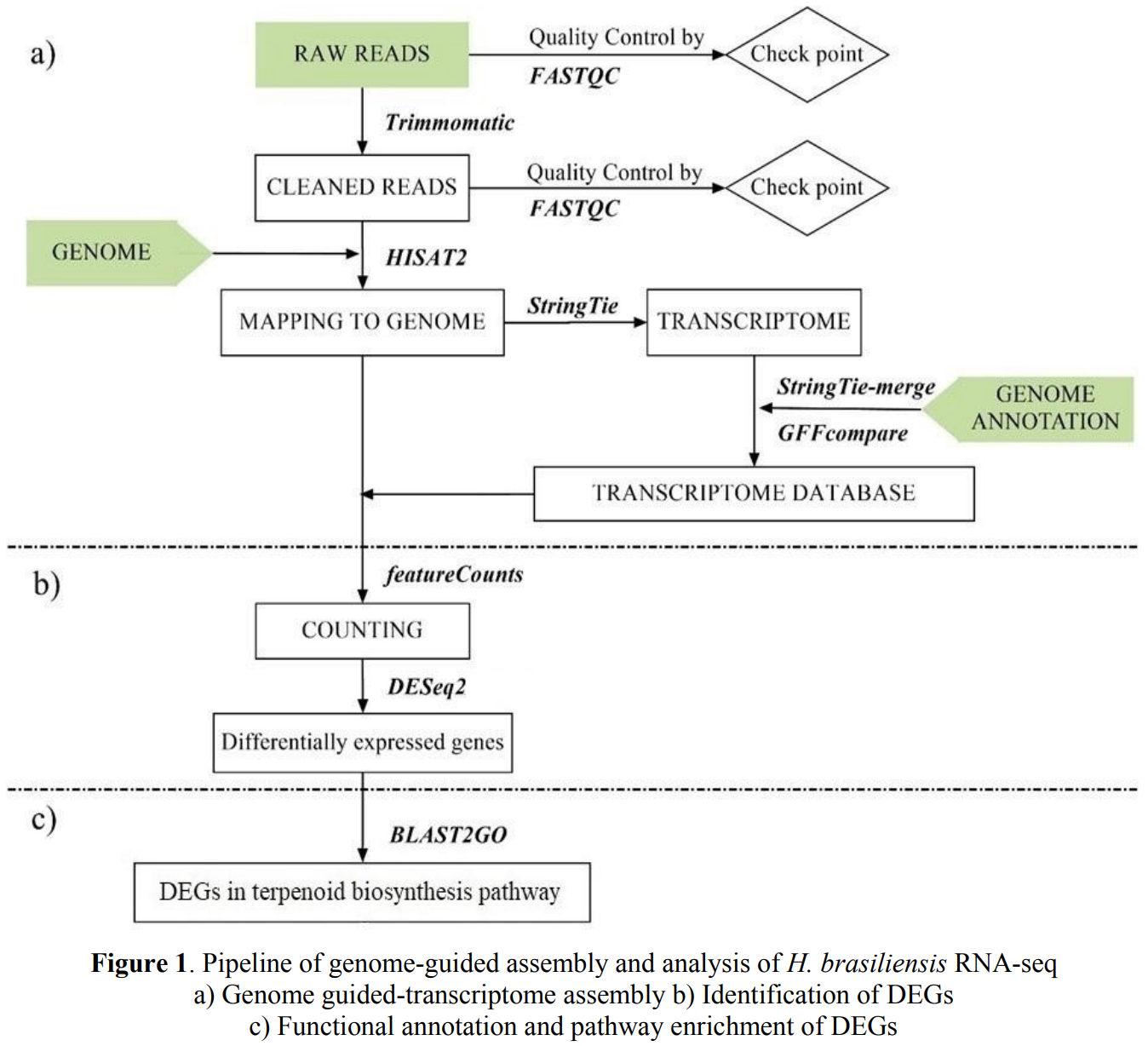
Transcriptomic Profiling, Functional Annotation, and Differentially Expressed Genes (DEGs)
Whole transcriptome sequencing (RNA-seq) provides critical insights into the molecular mechanisms that govern biological processes, making it an essential tool for biomedical research. By comparing treatment and control datasets, researchers can identify differentially expressed genes (DEGs), which helps to highlight how specific conditions or treatments influence gene expression in different cell types. This data can aid in diagnosing genetic diseases by comparing DEGs with known genetic markers, and can also help determine disease progression by quantifying expression changes linked to specific disease stages.
In one of my projects, I developed a pipeline to identify DEGs from RNA-sequencing data, as detailed in my publication (Van NT La et al., 2021). This pipeline allows for the comprehensive analysis of transcriptomic data, helping to identify significant gene expression changes under various experimental conditions. While this project primarily focused on Hevea brasiliensis (rubber tree), the pipeline is versatile and can easily be adapted for RNA-seq data analysis in other biological systems, including human health studies. For example, the same approach could be applied to understand molecular mechanisms in diseases like cancer, cardiovascular disorders, or infectious diseases.
Functional annotation then helps elucidate the molecular functions, biological processes, and subcellular locations of the genes. This provides a clearer understanding of how these genes contribute to the condition being studied. The pipeline integrates bioinformatics tools that can be employed for gene function annotation, helping researchers to infer biological pathways and potential therapeutic targets.
Analyzing DEGs with Machine Learning Methods
After identifying DEGs, the next step is to further analyze the results using machine learning techniques. Machine learning provides a powerful approach to classify cell types, identify key features, and predict outcomes based on gene expression data.
In other projects, I applied machine learning methods to classify human cell lines related to disease. Supervised learning techniques such as Linear Discriminant Analysis (LDA), Naive Bayes, Support Vector Machines (SVM), and Random Forest were employed to classify and predict disease-related patterns from DEGs. Unsupervised techniques like hierarchical clustering (H-clust), K-means, and Principal Component Analysis (PCA) were also used to explore inherent structures in the data. The application of these machine learning algorithms led to the identification of significant features and potential biomarkers, enhancing our understanding of disease mechanisms.
These methods, in combination with DEG identification, provide valuable insights into disease processes and potential therapeutic targets.
Genomic Analysis
Genomic analysis plays a crucial role in understanding genetic variations and their impact on disease. I worked on two distinct projects related to genomic data:
-
Copy Number Variant (CNV) Detection using Whole Exome Sequencing (WES)
I developed a pipeline for CNV detection using WES data. CNVs, which involve the duplication or deletion of sections of the genome, are linked to various genetic disorders and early-onset diseases. The pipeline I created provides an effective means of identifying these variants, offering clinicians insights into potential genetic risks.
-
Chromosomal Anomaly Identification using Whole Genome Sequencing (WGS)
I worked on identifying chromosomal anomalies, such as trisomies, using WGS data. This project focused on the detection of conditions like Down syndrome (trisomy 21), Edwards syndrome (trisomy 18), and Patau syndrome (trisomy 13). The WGS-based pipeline allows for early, non-invasive diagnosis of these anomalies, providing crucial information for prenatal care.
Summary
These projects highlight the power of sequencing technologies in various biomedical studies, ranging from disease research to the identification of genetic mutations and enabling early diagnosis.